Kayser-Fleischer Ring
🧠 Kayser–Fleischer Ring — Overview
Definition:
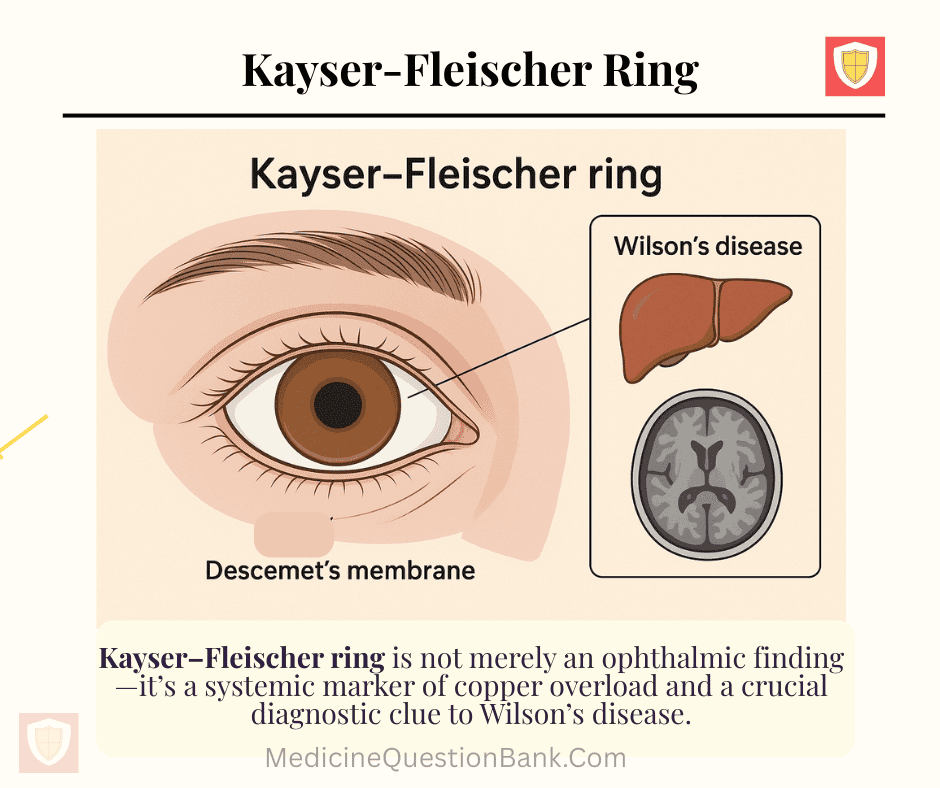
A Kayser–Fleischer ring is a brownish or golden-green discoloration encircling the cornea at Descemet’s membrane, caused by copper deposition, most classically seen in Wilson’s disease (hepatolenticular degeneration).
🔬 Pathophysiology
- Copper accumulation due to defective ATP7B gene → impaired copper excretion into bile.
- Excess copper gets deposited in:
- Liver → hepatitis, cirrhosis
- Basal ganglia → movement disorders
- Cornea → Kayser–Fleischer ring
- Kidneys, bones, pancreas
👁️ Ophthalmic Appearance
- Location: Peripheral cornea, at Descemet’s membrane (deep corneal layer).
- Color: Golden to greenish-brown ring, more prominent at superior and inferior poles initially, later forming a full circle.
- Detection:
- Slit-lamp examination is diagnostic.
- May be subtle or invisible on gross inspection.
🧪 Associated Investigations
| Test | Finding |
|---|---|
| Serum ceruloplasmin | ↓ (low) |
| 24-hour urinary copper | ↑ (elevated) |
| Liver biopsy (hepatic copper content) | ↑ |
| Genetic testing | ATP7B mutation |
🧍♂️ Clinical Significance
- Pathognomonic for Wilson’s disease, especially when accompanied by neurologic or hepatic symptoms.
- May also be seen (rarely) in:
- Cholestatic liver diseases
- Primary biliary cholangitis
- Chronic active hepatitis
💊 Treatment (of underlying Wilson’s disease)
| Drug | Mechanism | Note |
|---|---|---|
| Penicillamine | Chelates copper, ↑ urinary excretion | First-line (monitor for hypersensitivity) |
| Trientine | Alternative chelator | Fewer side effects |
| Zinc acetate | Blocks intestinal copper absorption | Maintenance therapy |
🧩 Related Terms
| Term | Explanation |
|---|---|
| Sunflower cataract | Copper deposition in the lens → radiating brown-green opacities |
| Wilson’s disease | Autosomal recessive copper metabolism disorder due to ATP7B mutation |
| Descemet’s membrane | Deep corneal layer where copper deposits |
| ATP7B gene | Encodes copper-transporting ATPase; defective in Wilson’s disease |
💬 Mnemonic
“Wilson’s CAN’T” — Copper Accumulates in Nerves, Tissues (including cornea → K–F ring)
📊 Summary Table
| Feature | Kayser–Fleischer Ring |
|---|---|
| Cause | Copper deposition in Descemet’s membrane |
| Seen in | Wilson’s disease (pathognomonic) |
| Detection | Slit-lamp examination |
| Color | Golden-brown / greenish |
| Location | Peripheral cornea |
| Associated sign | Sunflower cataract |
| Treatment | Copper chelation (Penicillamine, Trientine, Zinc) |
🧠 Key Point
The Kayser–Fleischer ring is not merely an ophthalmic finding—it’s a systemic marker of copper overload and a crucial diagnostic clue to Wilson’s disease.
Kayser-Fleischer ring, Wilson’s disease eye sign, corneal copper deposition, Descemet’s membrane, slit lamp finding, sunflower cataract, ATP7B mutation, Wilson disease diagnosis, hepatic degeneration, ophthalmic copper ring, penicillamine therapy
🧠 Kayser–Fleischer Ring — 20 MCQs (Basic/Clinical)
Explanation: Copper deposition in Descemet’s membrane produces the Kayser–Fleischer ring, classically seen in Wilson’s disease.
Explanation: A slit-lamp exam (biomicroscopy) allows magnified visualization of the cornea and Descemet’s membrane where copper deposits lie.
Explanation: Copper is deposited in Descemet’s membrane, the deep basement membrane of the corneal endothelium.
Explanation: Wilson’s disease (hepatolenticular degeneration) causes systemic copper overload leading to corneal deposition and K–F rings.
Explanation: ATP7B encodes a copper-transporting ATPase; mutations impair biliary copper excretion causing accumulation.
Explanation: Copper accumulates in liver, brain, cornea, kidneys and other tissues—but the lungs are not a typical site.
Explanation: K–F rings are highly suggestive of Wilson’s disease, especially in patients with neurologic or hepatic features; they are effectively pathognomonic in the appropriate clinical context.
Explanation: Penicillamine is a chelator that increases urinary copper excretion; trientine is an alternative and zinc reduces absorption (maintenance).
Explanation: Wilson’s disease typically shows decreased ceruloplasmin and elevated 24-hour urinary copper; hepatic copper measurement confirms diagnosis.
Explanation: Basal ganglia (putamen, globus pallidus) are commonly involved causing movement disorders, tremor, dystonia, and parkinsonism features.
Explanation: Sunflower cataract is due to copper deposition in the lens capsule; it is less common than the K–F ring but characteristic of Wilson’s disease.
Explanation: Serum ceruloplasmin is generally low in Wilson’s disease, though levels can be normal in some cases—hence it must be interpreted with other tests.
Explanation: Wilson’s disease is inherited in an autosomal recessive manner due to biallelic ATP7B mutations.
Explanation: Zinc induces metallothionein in enterocytes which binds copper and reduces absorption; it is commonly used for maintenance therapy.
Explanation: The ring appears golden-brown to greenish-brown, often more obvious superiorly and inferiorly before forming a complete circle.
Explanation: K–F rings often begin at the superior and inferior corneal periphery and later extend circumferentially.
Explanation: ATP7B dysfunction leads to impaired incorporation of copper into ceruloplasmin and reduced biliary copper excretion, causing accumulation.
Explanation: Psychiatric features include personality changes, depression, irritability, and sometimes cognitive decline; they can precede neurological signs.
Explanation: Liver biopsy with direct quantitative measurement of hepatic copper is definitive for diagnosis when noninvasive tests are inconclusive.
Explanation: K–F rings can be absent in patients with only hepatic disease (more common in neurologic disease). They are copper (not iron) deposits and may take months to years to fade after therapy.
Pseudo-Kayser-Fleischer (KF) ring
Pseudo-Kayser-Fleischer (KF) ring is a corneal ring caused by deposits of bilirubin, not copper, that appear in people with severe cholestatic jaundice. The ring is a yellow to yellowish-green hue found in the posterior corneal stroma, and it can be mistaken for a true KF ring, which is a hallmark sign of Wilson’s disease. The accurate diagnosis of pseudo-KF rings is crucial to prevent misdiagnosing a patient with Wilson’s disease.
Distinguishing pseudo-KF rings from true KF rings
The key difference between a pseudo-KF ring and a true KF ring lies in the substance that forms the deposit, its location in the cornea, and its response to treatment.
| Feature | Pseudo-Kayser-Fleischer (PKF) ring | True Kayser-Fleischer (KF) ring |
|---|---|---|
| Composition | Bilirubin deposits. | Copper deposits. |
| Associated condition | Caused by high serum bilirubin levels in non-Wilsonian liver diseases, such as viral hepatitis, primary biliary cholangitis, and autoimmune hepatitis. | Caused by excessive copper accumulation in Wilson’s disease, a genetic disorder. |
| Appearance | A smooth, yellow to yellowish-green hue. | A granular, golden-brown or greenish-brown ring. |
| Location | In the posterior corneal stroma. | In Descemet’s membrane, the innermost layer of the cornea. |
| Progression | Typically appear circumferentially. | Starts in the superior cornea, then the inferior cornea, and eventually forms a complete ring. |
| Response to treatment | Reversible, and fades or disappears completely as the patient’s jaundice resolves and bilirubin levels decrease. | Reversible with chelation therapy, but the rings disappear much more slowly. |
Differential diagnosis
In addition to true KF rings, other corneal rings can be mistaken for pseudo-KF rings and must be considered in a differential diagnosis.
- Fleischer rings: Caused by iron deposits in the basal epithelial cells of the cornea and are associated with keratoconus, a condition that causes the cornea to thin and change shape.
- Primary biliary cholangitis (PBC): This chronic liver condition can cause high copper levels and, in some cases, lead to the formation of corneal rings that can be mistaken for pseudo-KF rings.
🧠 Kayser–Fleischer Ring — 20 Clinical Vignettes (USMLE / NEET-SS style)
Explanation: Wilson’s disease (ATP7B defect) causes impaired biliary copper excretion; excess copper deposits in Descemet’s membrane producing Kayser–Fleischer rings and causes hepatic and neuropsychiatric features.
Explanation: Liver biopsy with quantitative hepatic copper (>250 µg/g dry weight is diagnostic threshold) is the most direct measure; ceruloplasmin and urinary copper help but can be equivocal.
Explanation: Basal ganglia (putamen, globus pallidus) involvement in Wilson’s disease produces parkinsonian features—rigidity, bradykinesia, tremor, dystonia.
Explanation: Ceruloplasmin may be normal in some Wilson’s cases; measure 24-hour urinary copper (usually elevated) and consider hepatic copper quantification or genetic testing.
Explanation: Neurologic worsening can occur with chelation due to mobilization of copper; management includes specialist review—often switching to trientine or adjusting therapy rather than abrupt cessation.
Explanation: Wilson’s disease typically presents with decreased ceruloplasmin, increased 24-hour urinary copper, and high hepatic copper on biopsy.
Explanation: ATP7B-related Wilson’s disease is autosomal recessive; siblings have 25% chance to be affected—family screening is recommended.
Explanation: KF rings are more common in neurologic Wilson’s; they can be absent in patients with only hepatic disease.
Explanation: Ceruloplasmin can be low for multiple reasons and may be normal in some Wilson’s patients; diagnostic evaluation requires integrating slit-lamp exam, urinary copper (repeat/after penicillamine), hepatic copper, and genetics.
Explanation: Liver transplantation replaces defective ATP7B function, corrects copper metabolism, and is indicated for fulminant hepatic failure or decompensated cirrhosis; neurologic symptoms may or may not improve.
Explanation: Psychiatric manifestations of Wilson’s often include personality changes, irritability, depression, and behavioral disturbance and can precede neurologic or hepatic signs.
Explanation: Arcus senilis is lipid deposition in stroma seen in older adults; KF ring is copper deposition in Descemet’s membrane—different etiology and patient demographics.
Explanation: Massive hepatic copper release can cause Coombs-negative hemolytic anemia due to oxidative RBC damage; this can accompany fulminant hepatic failure in Wilson’s.
Explanation: Pregnancy requires specialist management; many patients continue maintenance (often zinc) or carefully adjusted chelation under obstetric and hepatology guidance—abrupt cessation risks flares.
Explanation: KF rings may fade slowly over months to years with effective copper chelation or after liver transplant; they do not disappear immediately.
Explanation: A penicillamine challenge (measuring 24-hour urinary copper after administration) can raise urinary copper and help diagnose borderline cases—used selectively and with caution due to potential worsening of neuro symptoms.
Explanation: Wilson’s can show steatosis, acute or chronic hepatitis, focal necrosis, and cirrhosis with markedly increased hepatic copper—histology is variable and requires quantitative copper assay for diagnosis.
Explanation: Penicillamine can cause nephrotic syndrome (proteinuria), cytopenias, and autoimmune phenomena—regular monitoring of urine, CBC, and clinical status is required.
Explanation: KF rings can be absent in early or isolated hepatic Wilson’s disease; diagnosis is clinical and lab-based, not solely dependent on slit-lamp finding.
Explanation: The combination of KF ring, low ceruloplasmin, and elevated urinary copper is highly suggestive of Wilson’s disease; definitive confirmation may require hepatic copper quantification or genetic testing.
“`