Erythropoietin (ESA) Resistance — 50 FAQs
Erythropoietin (ESA) Resistance — 50 FAQs
- What is ESA resistance?
Failure to achieve or maintain target hemoglobin despite appropriate or escalating ESA doses. - What is the alternative term for ESA resistance?
ESA hyporesponsiveness. - In which population is ESA resistance most common?
CKD patients, especially those on maintenance dialysis. - What is the single most common cause of ESA resistance?
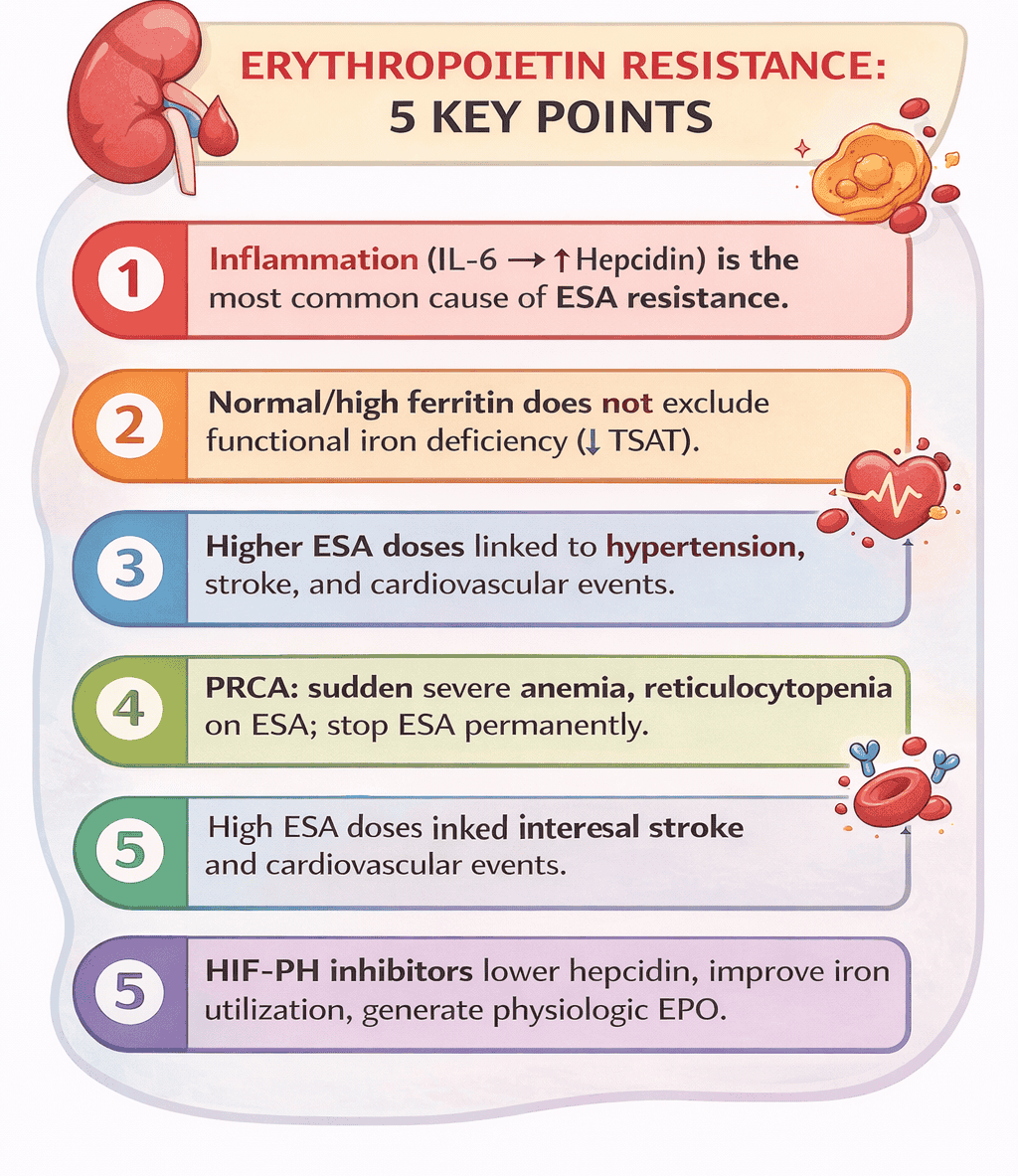
Chronic inflammation. - Which molecule is central to ESA resistance pathophysiology?
Hepcidin. - Which cytokine drives hepcidin production?
Interleukin-6 (IL-6). - What is functional iron deficiency?
Adequate iron stores but impaired iron availability for erythropoiesis. - How does functional iron deficiency present on labs?
Normal or high ferritin with low TSAT. - Why can ferritin be misleading in CKD?
It is an acute-phase reactant and rises with inflammation. - What defines absolute iron deficiency in CKD?
Low ferritin and low TSAT. - What iron route is preferred in ESA-resistant dialysis patients?
Intravenous iron. - Can IV iron be given when ferritin is high?
Yes, if TSAT is low and functional iron deficiency is present. - What is the best marker of ESA hyporesponsiveness severity?
ESA dose-to-hemoglobin ratio. - Why is aggressive ESA dose escalation discouraged?
It increases cardiovascular events, stroke, and hypertension. - Which CKD complication suppresses bone marrow erythropoiesis?
Secondary hyperparathyroidism. - How does high PTH cause ESA resistance?
By causing bone marrow fibrosis and erythroid suppression. - Which nutritional marker predicts poor ESA response?
Low serum albumin. - What does hypoalbuminemia reflect in CKD anemia?
Inflammation and malnutrition. - Which drugs blunt erythropoiesis?
ACE inhibitors and ARBs. - How do ACEi/ARBs impair erythropoiesis?
By inhibiting angiotensin II–mediated erythroid stimulation. - What dialysis factor contributes to ESA resistance?
Inadequate dialysis adequacy (low Kt/V). - Why does poor dialysis worsen anemia?
Uremic toxins suppress marrow response. - Which toxic exposure causes ESA resistance by impairing heme synthesis?
Aluminum. - Which vitamin deficiencies should be excluded?
Vitamin B12 and folate deficiency. - Why must occult blood loss be excluded first?
It is a common, correctable cause of apparent ESA failure. - Which laboratory test best reflects marrow response to ESA?
Reticulocyte count. - What does absent reticulocyte response suggest?
Bone marrow failure or PRCA. - What is pure red cell aplasia (PRCA)?
Antibody-mediated destruction of erythroid precursors due to anti-EPO antibodies. - How does PRCA present clinically?
Sudden severe anemia after prior ESA response. - What is the hallmark lab feature of PRCA?
Severe reticulocytopenia. - Are iron indices abnormal in PRCA?
No, they are typically normal. - What is the management of ESA-induced PRCA?
Permanent discontinuation of ESA and immunosuppression. - Why is ESA resistance a poor prognostic marker?
It reflects systemic inflammation and high cardiovascular risk. - Which outcomes are associated with high ESA doses?
Hypertension, stroke, vascular access thrombosis, mortality. - What hemoglobin target should be avoided in CKD?
Hb ≥13 g/dL. - Is transfusion first-line therapy for ESA resistance?
No, it is reserved for refractory or symptomatic anemia. - What is the role of HIF-PH inhibitors in ESA resistance?
They improve anemia by reducing hepcidin and enhancing iron utilization. - How do HIF-PH inhibitors differ from ESAs?
They produce near-physiologic endogenous EPO levels. - Why are HIF-PH inhibitors effective in inflammatory states?
They bypass hepcidin-mediated iron blockade. - Name examples of HIF-PH inhibitors.
Roxadustat, daprodustat, vadadustat. - What is the first step when Hb does not respond to ESA?
Reassess iron status and inflammation. - Should ESA dose be increased before correcting iron deficiency?
No. - What is the most frequent reversible cause of ESA resistance in dialysis units?
Catheter-related infection. - Which lab pattern suggests inflammation-driven anemia?
High ferritin with low TSAT and elevated CRP. - Why is ESA resistance not just a hematologic issue?
It reflects multisystem disease burden. - What principle guides ESA resistance management?
Treat the underlying cause, not the Hb number. - What is the earliest indicator of ESA effectiveness?
Rise in reticulocyte count. - Which factor shortens RBC lifespan in CKD?
Oxidative stress and uremic milieu. - Is ESA resistance more common in malnourished patients?
Yes. - One-line takeaway for exams?
ESA resistance = inflammation-driven iron restriction with high CV risk—correct the cause, not the dose.
ESA Resistance & HIF-PH Inhibitors — 40 Ultra–High-Difficulty SS MCQs
Q1. In CKD patients with ESA resistance, which pathway primarily explains reduced iron availability despite normal ferritin?
Q2. Which trial-level concern limited aggressive ESA dose escalation in CKD?
Q3. ESA hyporesponsiveness index is best expressed as:
Q4. Which mechanism allows HIF-PH inhibitors to remain effective during inflammation?
Q5. Compared with ESAs, HIF-PH inhibitors generate EPO levels that are:
Q6. Functional iron deficiency is best defined as:
Q7. Which ESA complication correlates most with dose intensity rather than Hb achieved?
Q8. Sudden loss of ESA response with near-zero reticulocytes suggests:
Q9. Which lab pattern most strongly suggests inflammation-driven ESA resistance?
Q10. Secondary hyperparathyroidism causes ESA resistance primarily via:
Q11. Which dialysis factor most strongly predicts ESA resistance?
Q12. In ESA-resistant CKD, IV iron is justified when:
Q13. Which medication class suppresses erythropoiesis via angiotensin II inhibition?
Q14. Aluminum-related ESA resistance is due to impaired:
Q15. Which outcome best explains why ESA resistance is prognostically adverse?
Q16. HIF-PH inhibitors increase iron availability by increasing:
Q17. Which ESA complication is dose-dependent rather than Hb-dependent?
Q18. Best initial step when Hb fails to rise after ESA initiation?
Q19. Which nutritional marker correlates best with ESA response?
Q20. Absolute iron deficiency differs from functional iron deficiency by:
Erythropoietin Resistance — 50 Ultra-Short One-Liners
- ESA resistance is defined by a high ESA dose requirement with poor hemoglobin response.
- ESA hyporesponsiveness is most common in CKD and dialysis patients.
- Inflammation is the commonest cause of ESA resistance.
- IL-6–driven hepcidin excess is central to ESA resistance.
- Hepcidin blocks ferroportin and traps iron in macrophages.
- Functional iron deficiency means iron is present but unavailable.
- Normal or high ferritin does not exclude iron deficiency in CKD.
- Low TSAT with normal or high ferritin indicates functional iron deficiency.
- Ferritin acts as an acute-phase reactant in CKD.
- IV iron can improve ESA response despite high ferritin levels.
- ESA dose-to-hemoglobin ratio best quantifies ESA resistance.
- High ESA doses predict increased cardiovascular risk.
- ESA dose intensity matters more than achieved hemoglobin.
- ESA trials showed higher stroke risk with higher Hb targets.
- ESA resistance is a poor prognostic marker.
- Secondary hyperparathyroidism suppresses marrow erythropoiesis.
- High PTH causes bone marrow fibrosis.
- Uremic toxins reduce erythroid progenitor responsiveness.
- Inadequate dialysis worsens ESA responsiveness.
- Hypoalbuminemia reflects inflammation and poor ESA response.
- ACE inhibitors and ARBs blunt erythropoiesis.
- Angiotensin II physiologically stimulates erythroid progenitors.
- Aluminum toxicity impairs heme synthesis.
- Absolute iron deficiency shows low ferritin and low TSAT.
- Occult blood loss must be excluded before escalating ESA.
- Reticulocyte count is the earliest marker of ESA response.
- Absent reticulocytosis suggests marrow failure or PRCA.
- Pure red cell aplasia presents with sudden ESA failure.
- PRCA is caused by anti-erythropoietin antibodies.
- In PRCA, iron indices are typically normal.
- ESA must be permanently discontinued in PRCA.
- Blood transfusion is not first-line therapy for ESA resistance.
- Target hemoglobin ≥13 g/dL should be avoided in CKD.
- ESA resistance reflects systemic disease rather than isolated anemia.
- Catheter-related infection is a frequent reversible cause of ESA resistance.
- Chronic inflammation shortens red cell survival.
- High ESA doses increase hypertension risk.
- Vascular access thrombosis correlates with ESA dose.
- HIF-PH inhibitors stabilize hypoxia-inducible factor.
- HIF-PH inhibitors reduce hepcidin levels.
- HIF-PH inhibitors improve iron mobilization.
- HIF-PH inhibitors produce near-physiologic endogenous EPO.
- HIF-PH inhibitors are effective in inflammatory anemia.
- Roxadustat is an oral HIF-PH inhibitor.
- Daprodustat is approved for CKD-related anemia in several regions.
- ESA resistance improves more with cause correction than dose escalation.
- Iron correction should precede ESA dose increase.
- ESA resistance increases mortality independent of hemoglobin level.
- Treating inflammation improves ESA responsiveness.
- The key principle in ESA resistance is to treat the cause, not the number.